Long-read Sequencing for Population-scale Genomic Study
Long-read Sequencing for Population-scale Genomic Study
Blog Article
Population genetics and precision health research rely on large genomic datasets. Long-read sequencing from Pacific Biosciences and Oxford Nanopore Technologies (ONT) has achieved a level of accuracy and throughput that allows for the progression from single genomes and small populations of individuals to the detection of variation in large-scale populations. Population-scale genomic studies are important, including reflecting the genetic diversity of target populations, detecting challenging genomic regions, serving as a resource for population genetics, translational research, and drug discovery, etc.
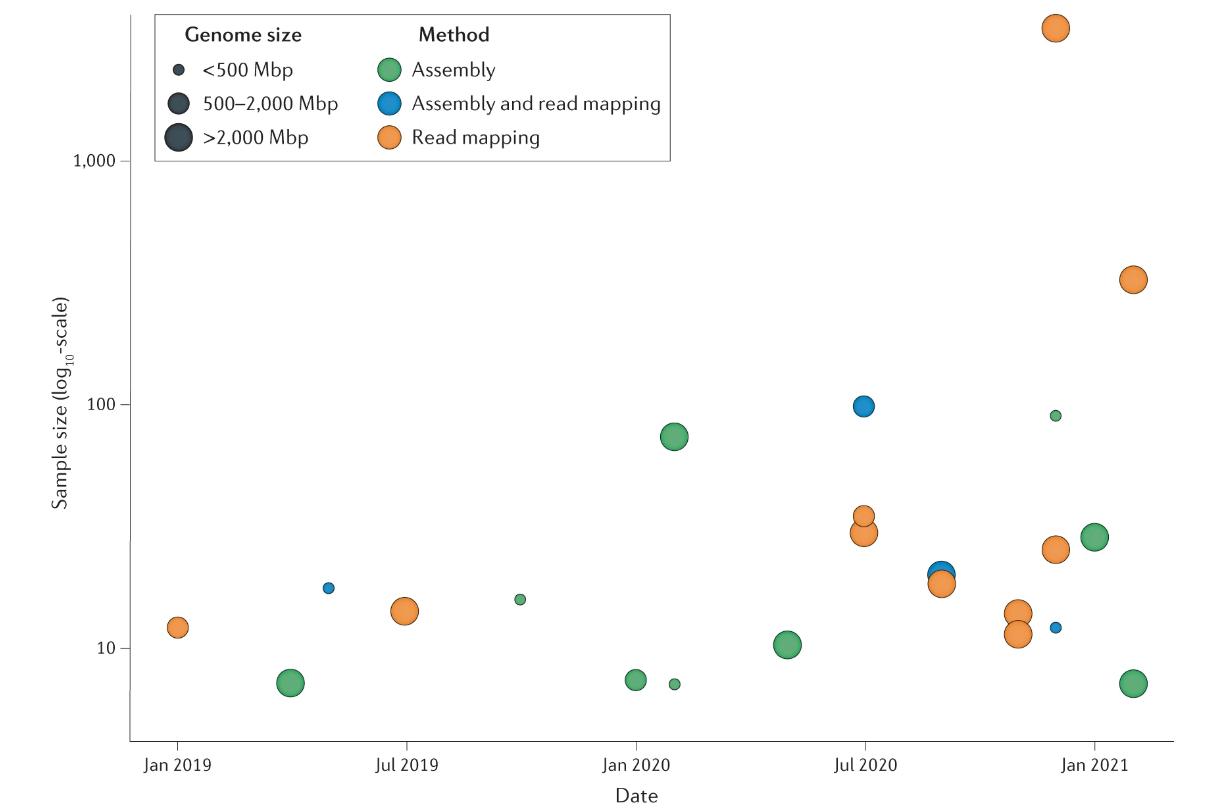
Overview of population-scale studies using long-read sequencing. (De Coster et al., 2021)
Overview
Sequencing the deoxyribonucleic acid (DNA) or messenger ribonucleic acid (mRNA) of different individuals in single or multispecies populations (known as population-scale sequencing) is fundamentally aimed at revealing allelic variation in macroscopic population profiles. This approach provides a critical scaffold for addressing multifaceted queries spanning the research fields of evolutionary biology, agronomic biotechnology, and translational medicine. Historical precedents of population-centric genomic studies, especially genome-wide association studies (GWAS), have always faced challenges in capturing the full range of genetic determinants of human phenotypic expression and pathological manifestations. This gap in understanding can largely be attributed to the intricate network of structural variation (SV). These SVs include inversions, deletions, and other complex chromosomal rearrangements that often remain elusive in the face of traditional sequencing methods.
High-throughput short-read sequencing platforms are characterized by read lengths that fluctuate between 25 base pairs (bp) and an upper limit of 400 bp. Their abilities are often hampered when they are tasked with deciphering variations hidden within the "dark matter" regions of the genome. Furthermore, they do not perform well in accurately resolving broad or complex variants. These obstacles not only compromise the integrity of genetic inferences derived from ancestry cohort datasets, but ultimately lead to a weakened, if not fragmented, understanding of the intricate interplay between genetic markers and disease etiology.
Emerging on this horizon is the promising field of long-read sequencing. This format enables the interrogation of genomic fragments spanning considerable contiguous lengths. The resulting capability is a holistic characterization of SVs across the human genomic landscape, setting the stage for an era dominated by population-scale long-read sequencing. By leveraging this cutting-edge technique, researchers are poised to unearth previously mysterious SVs with important links to phenotypic expression in humans, crops, fruit flies, and even birds such as songbirds. This paradigm shift is not just a technological advance, but marks a transformative leap in metagenomic research, heralding unprecedented insights and breakthroughs.
Project Strategies for Population-scale Sequencing
At the start of a population-scale sequencing project, there are multiple strategies with specific budget requirements to consider, as shown below. These strategies allow for different sizes and budgets, which can have an impact on the level of resolution at which genetic variants are detected.
Full Coverage Approach
This strategy is designed to sequence every sample from a population with moderate to high coverage, allowing for the highest level of resolution. The main criterion for determining the coverage required for each sample is whether it is assembled from scratch (requiring >40-fold coverage) or a reference-based comparison method (requiring >12-fold coverage42 ). The advantages of this strategy are its comprehensiveness, simplicity of study design, and relatively simple computational workflow. In addition, the samples are similarly covered and therefore equally well-studied, and rare variations in each sample can be easily detected.
Mixed Coverage Approach
In a "mixed-coverage" approach, a subset of samples representing subgroups (e.g., ethnicities or subgroups) of a cohort is sequenced at high coverage, and the remaining samples are sequenced at low coverage. Although this approach is generally less expensive than the full coverage approach, it still achieves higher overall detection sensitivity and is therefore particularly suitable for studies with a large number of individuals or a limited budget. However, some analytical challenges remain, especially in achieving high accuracy of genotypes across multiple samples or in distinguishing somatic versus heterozygous germline variants, which is further complicated by regions exhibiting recurrent mutations. In addition, this hybrid coverage approach will certainly bias against common alleles, as many rare alleles may be missed, especially when a locus is heterozygous and alternative alleles are therefore sparsely covered.
Hybrid Sequencing Methods
This approach involves sequencing only a small number of samples (e.g., 10-20% of all samples) with long reads, sequencing the remaining samples with short reads, and genotyping the variants found in the long reads. Once a subset of samples has been sequenced using the long read technique to produce a set of identified SVs, they can be genotyped for their breakpoint coordinates in the short read long sequence dataset. In this way, robust allele frequencies for the identified variants can be obtained. This strategy has been applied to diversity panels of human SVs to discover new expression quantitative trait loci (eQTL) and evolutionarily adapted traits.
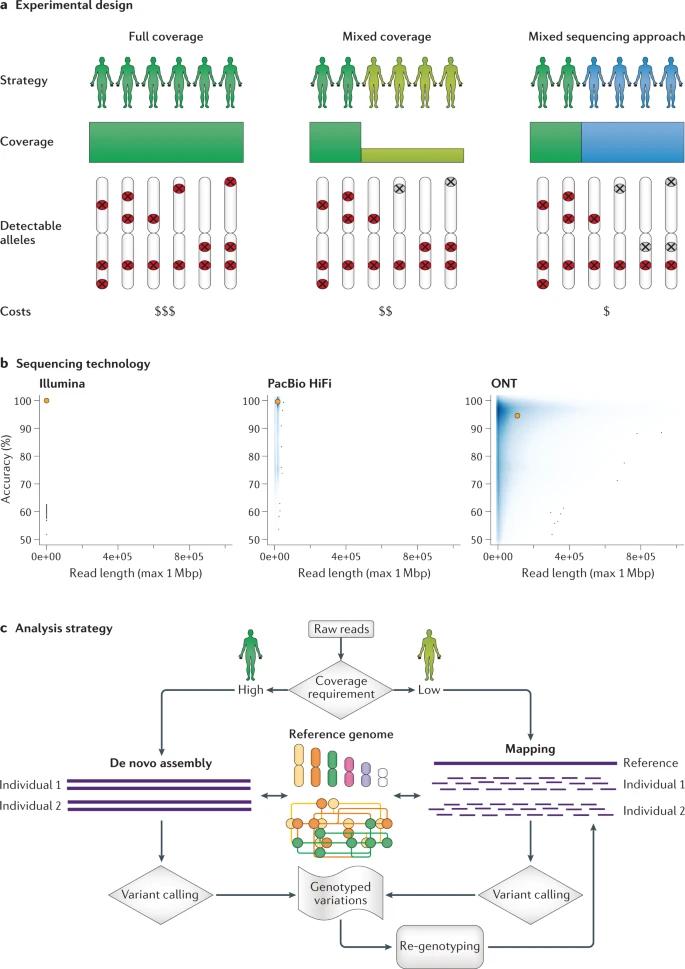
Overview of long-read population study design. (De Coster et al., 2021)
The Importance of Long-Read Sequencing Technology in Population-Scale Study
One of the inherent challenges of population genetics is the accurate phasing of haplotypes-determining specific combinations of alleles located on a single chromosome. Long-read sequencing provides an effective solution by capturing longer DNA fragments, which can directly determine haplotype structure without relying on computational prediction or family-based studies. This capability is transformative for population-scale studies, where understanding the distribution and combination of specific allele sets can help decipher population history, migration patterns, and shared inheritance patterns.
Structural variants, such as deletions, duplications, and inversions, can have profound effects on gene function and expression. Capturing these variants at high resolution is critical when studying large populations. Long-read sequencing can identify structural variants that may be overlooked or inaccurately represented by short-read methods.
Population-scale Sequencing Downstream Analysis Methods
The choice of analytical tools is critical for downstream analysis at the population scale. Prior to downstream analysis, quality control must be performed on experimental factors that directly affect the performance of assembly, SV detection, and read-sequencing phases. There are several strategies for population-scale downstream analysis:
Read Alignment-based Analysis
Comparison-based methods are often the preferred approach for population-scale studies because they facilitate the comparison of all samples to a common coordinate system (i.e., the reference genome). In addition, these methods are usually less computationally demanding and require much less coverage than compilation-based methods. Comparison-based methods rely on matching sequencing reads to a reference genome, the overall correctness of which will affect the analysis of the read data.
Software for analyzing long-read sequence data, such as NGMLR and LAST methods, speeds up the matching process and improves the accuracy of long-read matching. In addition, a variety of tools for detecting genetic variation can eliminate the need for high sequencing coverage by enabling SV calling and genotyping at lower coverage.
Population-scale De Novo Assemblies
Traditional reference genomes, often based on short-read sequencing, can be fragmented and may miss key sequences. Such omissions may lead to significant differences, including false-positive or false-negative variant identifications. Therefore, there is an urgent need to construct and compare scratch assemblies.
The increased availability and affordability of long-read sequencing data have led to an explosion of faster and more accurate genome assembly tools. De novo assembly-based methods are often more sensitive and better suited to reconstructing highly diverse regions of the genome than comparison-based methods. The increasing yield of long-read sequencing technologies will allow sufficient coverage of each sample to be sequenced for high-quality de novo assembly.
Graph Genome Methods
Both read matching and de novo assembly methods can have systematic problems with complex structural variants, missing insertion sequences in the reference genome, repetitive variants, and highly polymorphic loci. A major benefit of graph genomes is the use of short reads for genotyping SVs. In addition, with this graph-based approach, for population studies, the often discussed dichotomy of using an existing reference genome for alignment or constructing a new reference genome by assembling it from scratch can be avoided since downstream of this step all sequences have to be aligned with the backbone of the individual (reference) assembly or pan-genome map for identification of variants, annotation, and statistical evaluation.
References
- De Coster, Wouter, Matthias H. Weissensteiner, and Fritz J. Sedlazeck. "Towards population-scale long-read sequencing." Nature Reviews Genetics 22.9 (2021): 572-587.
- Rech, Gabriel E., et al. "Population-scale long-read sequencing uncovers transposable elements associated with gene expression variation and adaptive signatures in Drosophila." Nature Communications 13.1 (2022): 1948.